



 – Dieser Artikel entstand in Zusammenarbeit mit Minitab Area Marketing Manager Abbie Wong.
– Dieser Artikel entstand in Zusammenarbeit mit Minitab Area Marketing Manager Abbie Wong.
Als es durch COVID-19 weltweit zu Engpässen bei medizinischen Produkten kam, bemühten sich viele Hersteller, ihre Produktionslinien zu optimieren, um der Nachfrage gerecht zu werden. Auch wenn die Food and Drug Administration (FDA) in den USA die Richtlinien für einige Produkte wie Handdesinfektionsmittel vorübergehend lockerte, gilt doch ein Standardweg für die Zulassung, an den sich die Hersteller bei der Markteinführung ihrer Produkte halten müssen.
Wir betrachten hier die fünf Phasen, die die FDA empfiehlt, sowie einige Beispiele für die statistischen Verfahren, die dabei häufig zum Einsatz kommen.
PHASE 1: BEDARF UND KONZEPT
Sie kennen sicher den Satz „Not macht erfinderisch“. Wie bei vielen neuen Produkten und Innovationen ganz allgemein beginnen die Hersteller von medizinischen Produkten häufig mit einem Projekt, das dann die FDA-Phasen durchläuft, weil sie einen Bedarf erkennen. Ob Herzschrittmacher oder eine Kamera in einer Pille: Für alle medizinischen Produkte wird zuerst ein Konzeptnachweis benötigt. Hiermit belegen die Entwickler, dass ihr Konzept einen praktischen Nutzen hat.
In einigen Fällen gibt es mehrere ähnliche Produkte oder ein Produkt, für das andere Materialien verwendet werden. Daten zu Zuverlässigkeit, Einheitlichkeit und vielen anderen Faktoren können in einer Streuungskarte in Minitab dargestellt werden, um zu bestimmen, welches Produkt am besten für die Weiterentwicklung geeignet ist.
Hier finden Sie dieses und andere Beispiele aus der Praxis. Sehen Sie sich unsere Webinar-Aufzeichnung in englischer Sprache Statistical Solutions to Help You with the 5 FDA Medical Devices Stages an >

PHASE 2: DESIGN UND PROTOTYP
In dieser Phase erstellen die Entwickler eine erste Version eines medizinischen Produkts – nicht für den Einsatz am Patienten, sondern zum Testen unter kontrollierten Laborbedingungen. Die Optimierung des Prototyps liefert Informationen zum möglichen Einsatz des Produkts in der Praxis und dazu, wie Risiken minimiert werden können.
Beispiel: Lebenszyklus eines Produkts mit einer beschleunigten Lebensdauerprüfung prognostizieren
Einige Produkte, z. B. implantierbare Geräte, haben eine Lebensdauer von vielen Jahren. Daher muss jedes medizinische Produkt mit einem Ablaufdatum versehen werden, das durch Daten zur Haltbarkeit belegt wird.
Um Daten zu Ausfällen zu erfassen, können die Entwickler die Verfahren für die beschleunigte Lebensdauerprüfung (ALT) in der Minitab Statistical Software einsetzen. Hierbei wird der Ausfall eines Produkts unter extremen Bedingungen provoziert, z. B. bei hohen Temperaturen oder starkem Druck. Dadurch wird die erforderliche Testzeit reduziert. Die Fehlerdaten werden dann analysiert, um den Produktlebenszyklus unter normalen Bedingungen zu extrapolieren.
Steht Ihre Organisation vor Herausforderungen bei der Zuverlässigkeit? Laden Sie unser White Paper herunter, um die 5 Methoden kennenzulernen, durch die Sie mit Hilfe der Minitab Solutions Analytics eine zuverlässige Produktdesign entwickeln.

PHASE 3: DER WEG ZUR ZULASSUNG
Der Weg zur Zulassung eines medizinischen Produkts hängt von seiner Risikoklasse ab (Klasse I, II oder III). Jedes Produkt wird abhängig davon klassifiziert, wie intensiv es kontrolliert werden muss, um seine Sicherheit und Wirksamkeit mit hinreichender Zuverlässigkeit bestätigen zu können. Auf der Website der FDA finden sich ausführliche Beschreibungen zur Klassifizierung von Produkten sowie zu den jeweils erforderlichen Tests und Bestätigungen. Einfach ausgedrückt fallen in Klasse I einfache Produkte wie Verbände, Latexhandschuhe und elektrische Zahnbürsten, während es sich bei Klasse III im Allgemeinen um lebenserhaltende Geräte handelt, z. B. die implantierbaren Geräte aus dem Beispiel oben.
In dieser Phase wird eine Charge des Produkts produziert und getestet, um die Leistung anhand der Spezifikationen zu beurteilen.
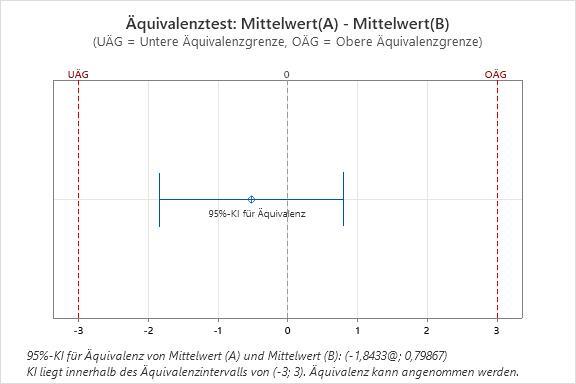
Beispiel: Äquivalenztests zum Bestätigen, dass zwei Produkte identisch sind
Mit statistischen Äquivalenztests kann beurteilt werden, ob zwei medizinische Produkte identisch sind. Äquivalenztests sind ein Analyseverfahren, mit dem eine Äquivalenz belegt wird. Hierbei werden die herkömmliche Null- und Alternativhypothese umgekehrt, d. h. als Nullhypothese wird angenommen, dass zwei Produkte nicht äquivalent sind und ein großer Unterschied zwischen ihnen besteht. Die Alternativhypothese ist, dass die Produkte identisch sind. So könnten Entwickler z. B. bestimmen, ob zwei Geräte für intravenöse Zugänge dieselbe Menge an Flüssigkeit abgeben. Sie definieren den Bereich der wissenschaftlichen Äquivalenz als mittlere Differenz bei der Infusionsmenge von höchstens 3 ml. Laut dem Äquivalenztest in Minitab können sich die Entwickler zu 95 % sicher sein, dass die Differenz zwischen der mittleren Infusionsmenge zwischen –1,84334 und 0,798674 ml liegt. Da das 95 %-Konfidenzintervall zwischen –3 und 3 liegt (Bereich der wissenschaftlichen Äquivalenz), sind die beiden IV-Geräte im Hinblick auf die Infusionsmengen äquivalent:

PHASE 4: PRÜFUNG DURCH FDA UND EINFÜHRUNG
In dieser Phase wird der Antrag auf Zulassung durch die FDA vorbereitet, damit das Produkt vermarktet werden kann. Dabei soll gezeigt werden, dass ausreichende und aussagekräftige Informationen zur Sicherheit und Wirksamkeit des Produkts vorliegen. Der Prozess wird überwacht, um sicherzustellen, dass er stabil ist. Dabei wird insbesondere darauf geachtet, dass das Produkt innerhalb der Spezifikationsgrenzen liegt.
One way to improve a medical device manufacturing process is to implement a statistische Prozesskontrolle (SPC). SPC-Programme werden normalerweise in der Massenproduktion eingesetzt, um eine fortlaufende Produktion zu ermöglichen. Hierbei werden Regelkarten verwendet, d. h. spezielle Zeitreihendiagramme, die abnormale Streuungsmuster in einem Prozess aufzeigen. So müssen nicht einzelne Chargen eines Produkts analysiert werden.
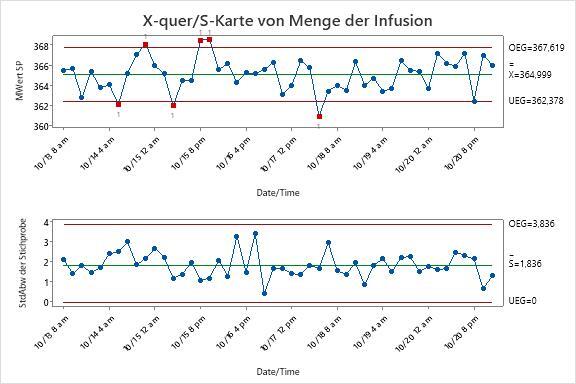
Beispiel: Abweichungen bei den Infusionsmengen von IV-Geräten prüfen
Ungefähr eine Woche lang werden fünf Geräte für den intravenösen Zugang zufällig aus jedem Los ausgewählt. Die Infusionsmenge wird gemessen. Aus den unten abgebildeten Regelkarten können die Tester entnehmen, welche Chargen außer Kontrolle sind. In den entsprechenden Protokolldateien können sie die Streuung durch Ausnahmebedingungen identifizieren und eliminieren. Wie unten dargestellt, können Hersteller mit einem erfolgreichen SPC-Programm stabile Prozesse sicherstellen, die Effizienz steigern und Kosten senken.
PHASE 5: SICHERHEITSPRÜFUNG NACH DER MARKTEINFÜHRUNG
Nachdem ein Produkt eingeführt wurde, wird die Produktionslinie überwacht, um sicherzustellen, dass sie ordnungsgemäß gewartet wird und der Prozess stets unter Kontrolle ist. In dieser Phase der Überwachung nach der Markteinführung muss außerdem gewährleistet werden, dass alle Vorfälle wie Ausfälle oder Fehlfunktionen gemeldet und behoben werden. Die FDA überprüft die Hersteller und bietet Programme an, über die Hersteller, medizinisches Personal und Kunden Probleme melden können.
Beispiel: Zeitplan für die vorbeugende Instandhaltung mit einer Zuverlässigkeitsanalyse aufstellen
In dieser Phase soll sichergestellt werden, dass die Maschinen bzw. Geräte im Werk angemessen funktionieren und es nur zu minimalen Ausfallzeiten kommt. Eine Form der Wartung ist die vorbeugende Instandhaltung, d. h. ein Vorgehen nach Zeitplan. Die vorbeugende Instandhaltung wird entsprechend der Risikobewertung eines medizinischen Produkts zu unterschiedlichen Zeitpunkten geplant. Mit den Zuverlässigkeitsstudien zur Haltbarkeit und der Analyse mehrerer Ausfallursachen in der Minitab Statistiksoftware können Hersteller das Ausfallrisiko in verschiedenen Phasen des Lebenszyklus für das gesamte Produkt und einzelne Komponenten berechnen. So kann die Wartung entsprechend dem Ausfallrisiko zu einem bestimmten Zeitpunkt im Lebenszyklus geplant werden.
ZUM ABSCHLUSS
Die Hersteller von medizinischen Produkten müssen die Qualität und Haltbarkeit in jeder der fünf Phasen des Produktlebenszyklus sicherstellen und genau dokumentieren. Wir hoffen, dass diese Beispiele mit den vielfältigen Analysewerkzeugen in der Lösungen von Minitab Ihnen zeigen konnten, wie der Produktlebenszyklus aufgebaut ist und durchlaufen wir.
Weitere Informationen: Ausführliche Details zu den 5 FDA-Phasen
