



 – Este blog foi escrito em parceria com a Gerente de Marketing de Área da Minitab, Abbie Wong.
– Este blog foi escrito em parceria com a Gerente de Marketing de Área da Minitab, Abbie Wong.
Quando os efeitos da COVID-19 causaram a escassez de dispositivos médicos em todo o mundo, muitos fabricantes correram para modificar suas linhas de produção de forma a atender à demanda. Apesar da Food and Drug Administration (FDA) dos Estados Unidos ter flexibilizado temporariamente as diretrizes para alguns produtos, como o desinfetante para as mãos, ainda há um caminho padrão para aprovação que os fabricantes de dispositivos médicos seguem a fim de comercializar seus produtos no mercado.
ESTÁGIO 1: DESCOBERTA E DESENVOLVIMENTO DO CONCEITO DE DISPOSITIVO
Você provavelmente já ouviu falar que a necessidade é a mãe da invenção. Como acontece com muitos novos produtos e inovações em geral, os fabricantes de dispositivos médicos iniciam um projeto e o desenvolvem de acordo com as metas das fases da FDA, porque perceberam uma necessidade a ser atendida. De um marca-passo até uma pílula com uma câmera dentro, todos os dispositivos médicos começam com uma prova de conceito, onde engenheiros e técnicos de P&D verificam se o conceito tem praticidade.
Às vezes, podem existir alguns produtos semelhantes (ou talvez iguais, mas usando materiais diferentes). Você pode conectar seus dados sobre confiabilidade, consistência e muitos outros fatores no Gráfico de variabilidade do Minitab para ajudar a decidir qual é o melhor produto para continuar o processo.
ESTÁGIO 2: CONCEPÇÃO E PROTÓTIPO
Nesta fase, os pesquisadores constroem uma versão inicial de um dispositivo médico - não para uso humano, mas para testar em ambientes controlados de laboratório. À medida que eles refinam o protótipo, continuam a aprender mais sobre o uso potencial do produto para as pessoas e como reduzir o risco de danos.
Exemplo: Previsão do ciclo de vida do produto usando Teste de vida acelerado
Alguns produtos, como dispositivos médicos para implantes, tendem a durar muitos anos. Por esse motivo, todo dispositivo médico deve ser rotulado com uma data de validade que seja confirmada por dados de tempo de vida.
Para coletar dados de falha, os pesquisadores podem aplicar as técnicas deTeste de Vida Acelerado (ALT) do Minitab. O ALT é obtido com um produto forçado a falhar mais rapidamente sob condições extremas, como alta temperatura ou alta pressão. Forçar o produto a falhar mais rapidamente reduz o tempo de teste necessário. Em seguida, os dados de falha são analisados para extrapolar o ciclo de vida do produto em circunstâncias normais.
ESTÁGIO 3: CAMINHO PARA A APROVAÇÃO
O caminho para a aprovação de um dispositivo médico depende de sua classificação de risco (Classe I, Classe II ou Classe III). Cada dispositivo recebe sua classe com base no nível de controle necessário para proporcionar uma garantia razoável de sua segurança e eficácia. O site da FDA tem descrições detalhadas sobre como os dispositivos são classificados e os testes e validação necessários para cada classe, mas para ilustrar de forma simples - a Classe I pode ser dispositivos simples como bandagens, luvas de látex e escovas de dente elétricas, enquanto a Classe III em geral abrange os que sustentam a vida, como os dispositivos médicos para implante do exemplo anterior.
Nesta fase, um lote de produtos é fabricado e testado para verificar seu desempenho em relação às especificações.
Exemplo: Confirmação de que 2 dispositivos são equivalentes usando o Teste de equivalência
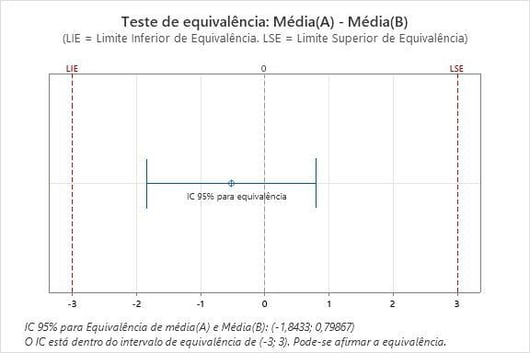
O Teste de equivalência estatística pode ser usado para avaliar se dois dispositivos médicos são equivalentes. O Teste de equivalência é uma abordagem analítica para proporcionar evidências de equivalência. Ao realizar um Teste de equivalência, as hipóteses nula e alternativa tradicionais são invertidas, o que significa que a hipótese nula é que dois dispositivos não são equivalentes (ou seja, a diferença entre eles é grande). A hipótese alternativa é que eles são os mesmos. Por exemplo, os pesquisadores desejam determinar se dois tipos de dispositivos de acesso intravenoso fornecem uma quantidade de fluido equivalente. Eles definem a zona de equivalência científica como uma diferença média na quantidade de infusão de 3 ml ou menos. Com o Teste de equivalência do Minitab, os pesquisadores podem ter 95% de certeza de que a diferença na quantidade média de infusão está entre -1,84334 e 0,798674 ml. Como o Intervalo de confiança de 95% está entre -3 e 3 (a zona de equivalência científica), os dois dispositivos IV são equivalentes para as quantidades de infusão:

ESTÁGIO 4: ANÁLISE PELA FDA E IMPLANTAÇÃO
Nesta fase, estamos nos preparando para apresentar um requerimento à FDA para comercializar o dispositivo para o público. Queremos demonstrar que temos informações suficientes e aceitáveis sobre a segurança e eficácia do dispositivo. Nós monitoramos o processo para garantir que seja estável e verificamos se estamos fabricando o produto dentro dos limites das especificações.
Uma maneira de melhorar o processo de fabricação de um dispositivo médico é implementar um programa de controle estatístico de processos (SPC). Normalmente usado em produção em massa, um programa SPC permite que uma empresa produza continuamente um produto usando gráficos de controle, um gráfico de séries temporais especializado que tenha sido criado para ajudar a identificar padrões anormais de variabilidade em um processo, em vez de inspecionar lotes individuais de um produto.
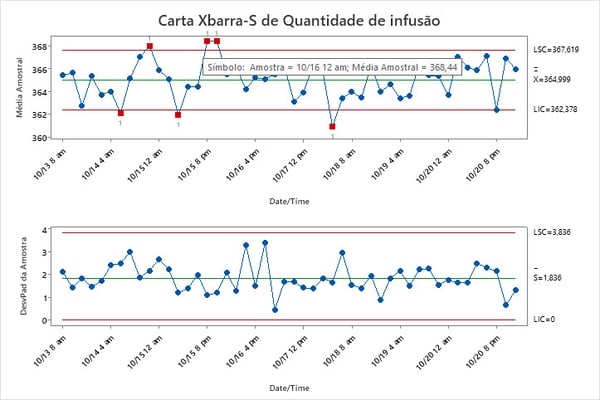
Exemplo: Verificação de possíveis alterações nas quantidades de infusão do dispositivo de acesso intravenoso
Cinco dispositivos intravenosos são selecionados aleatoriamente de cada lote ao longo de, aproximadamente, uma semana. A quantidade de infusão é medida. Nos gráficos de controle abaixo, os engenheiros podem identificar quais lotes estão fora de controle. Ao verificar os arquivos de registro correspondentes, eles podem identificar e eliminar todas as variações de causa especiais. Um programa SPC bem-sucedido ajuda os fabricantes a manter os processos estáveis, melhorar a eficiência e reduzir custos, conforme mostrado abaixo.
ESTÁGIO 5: MONITORAMENTO DE SEGURANÇA PÓS-COMERCIALIZAÇÃO
Agora que o produto foi lançado, vamos monitorar a linha de produção de forma correta para garantir que ela seja mantida adequadamente e que o processo permaneça em um estado de controle constante. Nessa fase de vigilância pós-comercialização, nós também garantimos que quaisquer eventos adversos, como falhas ou mau funcionamento do dispositivo, sejam relatados e tratados. A FDA realiza inspeções de fabricantes e emprega programas de relatórios que permitem que fabricantes, profissionais de saúde e consumidores relatem problemas.
Exemplo: Criar um cronograma de manutenção preventiva usando análise de confiabilidade
Esta fase envolve o fornecimento de um nível adequado de serviço e a limitação do tempo de inatividade dos dispositivos na instalação. Uma forma de manutenção é a manutenção preventiva (PM), que é um evento programado. As PMs são programadas de acordo com a classificação de risco do dispositivo médico em momentos diferentes. Os Estudos de vida útil via confiabilidade do Minitab e a Análise de modos de falhas múltiplas ajudam os fabricantes a calcular o risco de falha em um instante diferente do ciclo de vida do produto inteiro e de cada componente do produto. Os engenheiros podem então criar o cronograma de manutenção de acordo com a probabilidade de falha em diferentes estágios do ciclo de vida.
RESUMINDO
Os fabricantes de dispositivos médicos devem garantir a qualidade e durabilidade em cada uma das cinco fases do ciclo de vida do produto e documentá-las detalhadamente. Esperamos que esses exemplos da ampla variedade de ferramentas analíticas do Minitab possam lhe ajudar a entender e passar por cada fase do ciclo de vida do produto.
